論文が出版されました.
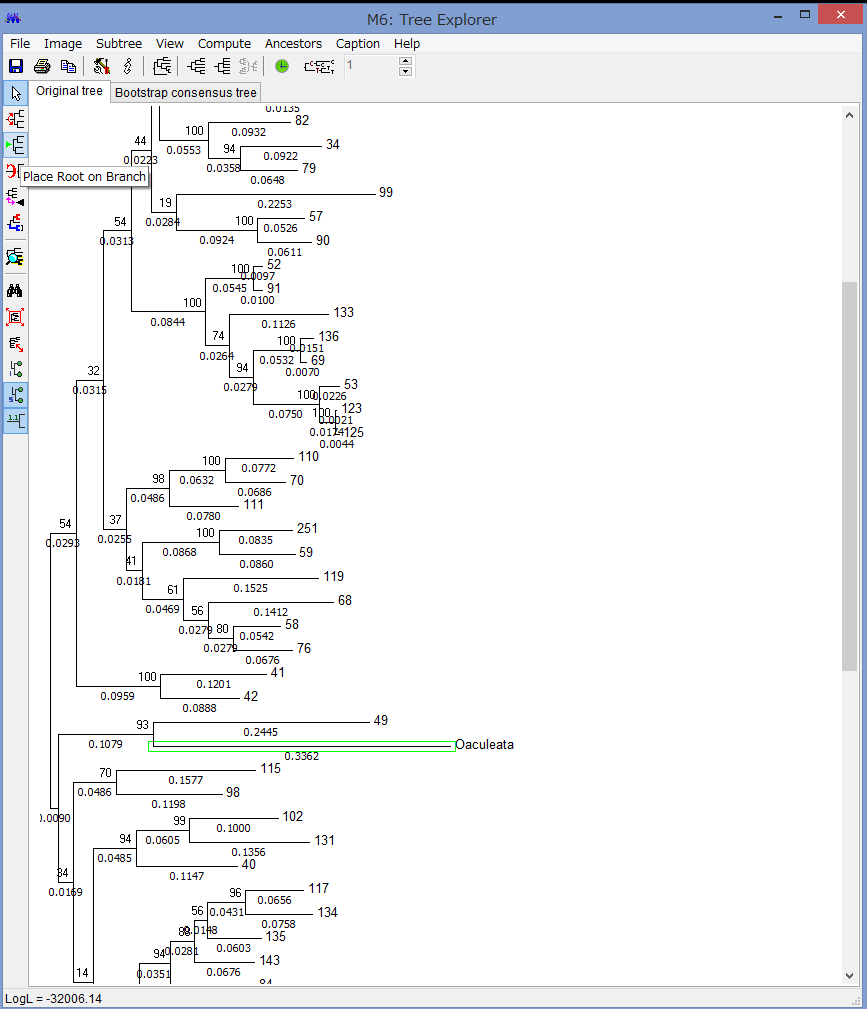
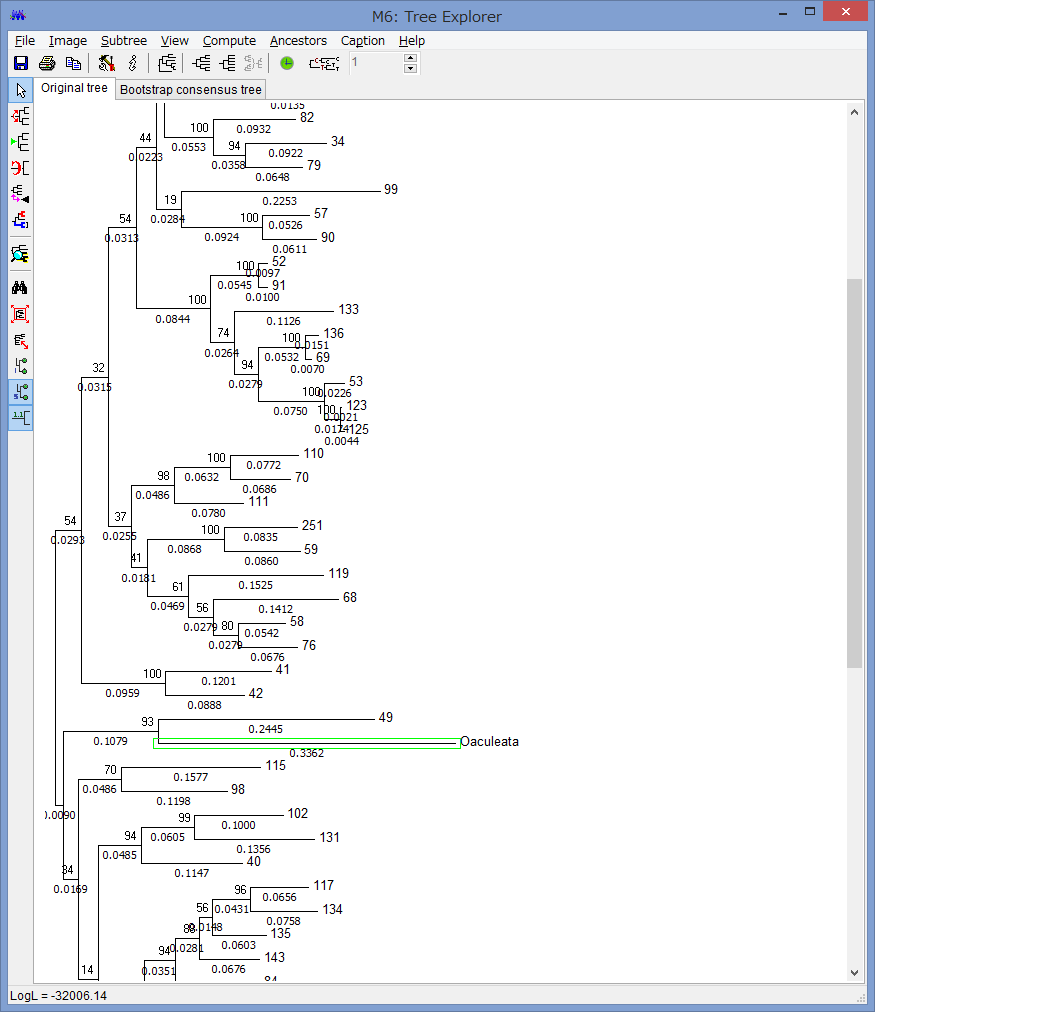
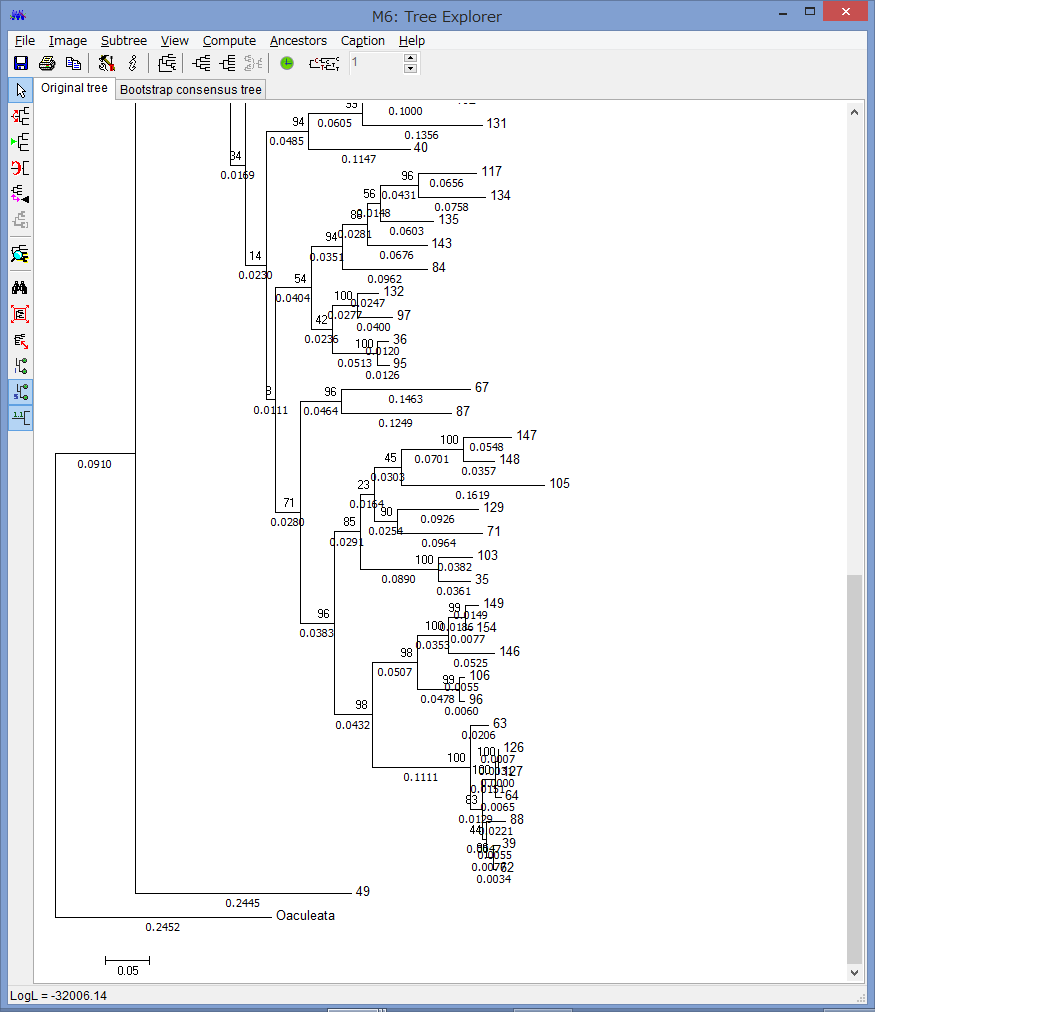
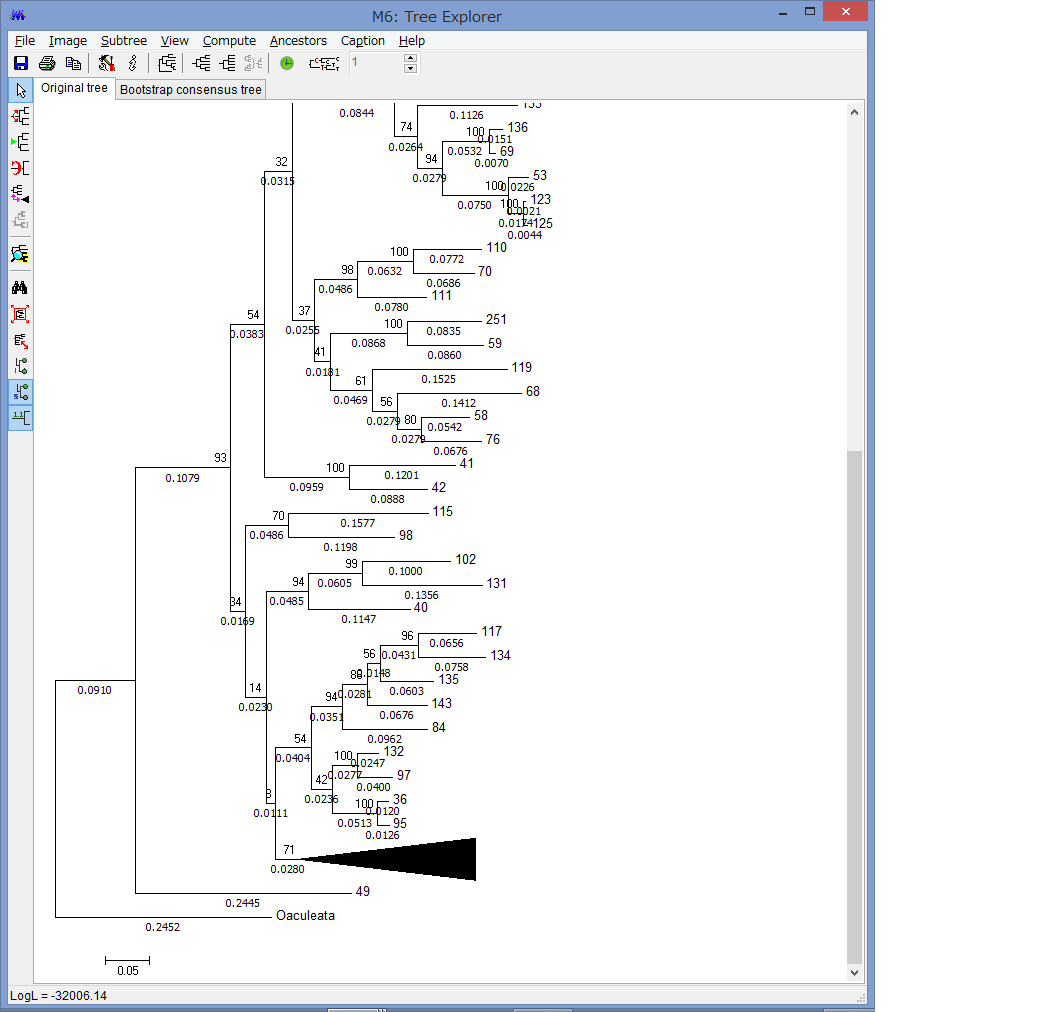
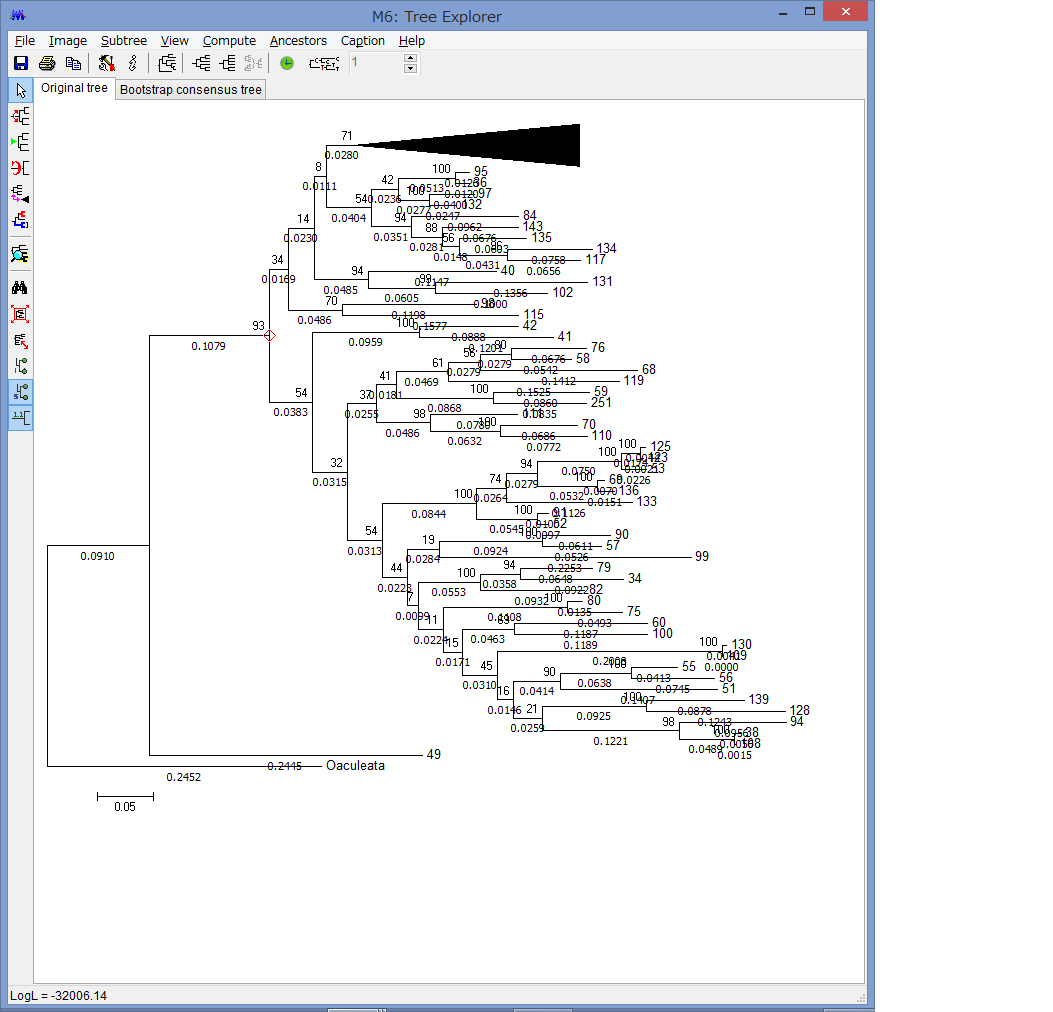
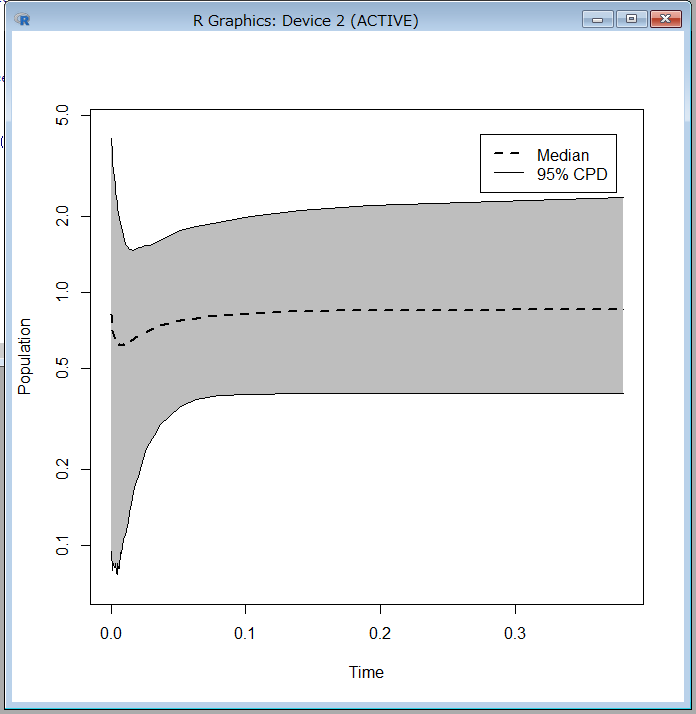
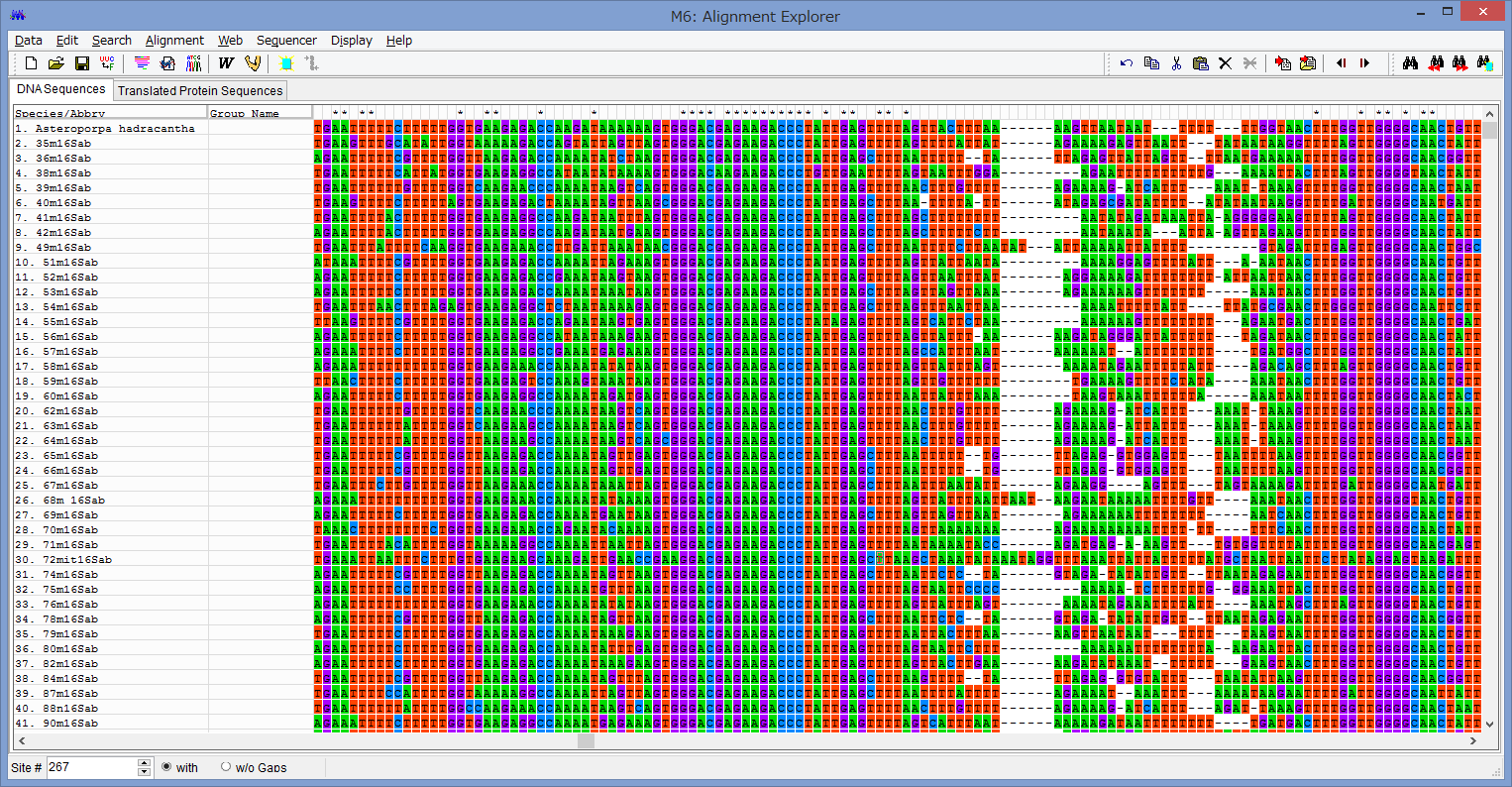
腕の先っちょだけが分岐する,サキワレテヅルモヅル属(Astroclon)という珍しい属の分子系統解析を行ったところ,同科の他のいかなるグループとも遠縁であることが分かりました.また,主要な近縁種の形態観察から,これまでに注目されてこなかった形態によっても他の属と区別できるため,本属は独立した亜科とする事が妥当,と結論し,新亜科Astrocloninaeを記載しました.
Okanishi, M*, Fujita, T. (2018)
“Description of a New Subfamily, Astrocloninae (Ophiuroidea: Euryalida: Gorgonocephalidae), Based on Molecular Phylogeny and Morphological Observations”.
Zoological Science. 35(2): 179—187.
Linkはコチラ
本研究で扱ったサキワレテヅルモヅルは,以前マイナビニュースで取り上げてもらった「分類学者が選ぶ! 好きなテヅルモヅ ルベスト3」の2位にランクインさせてもらったグループです.変わった腕の分岐パターンだけでなく,体が非常に頑健な骨片に囲まれている事もポイントです.
https://news.mynavi.jp/article/scientist_best3-3/
さらに,サキワレテヅルモヅル属の中の奇種中の奇種(といっても2種しかいない),イボサキワレテヅルモヅル(Astroclon suensoni)が,Zoological Scienceの表紙に選ばれました!好きなモヅルを論文にできて,しかも表紙に載せてあげることができてうれしいです.博士からの研究の続きではありますが,これでツルクモヒトデ目の系統に関する研究は一旦区切りになるかもしれません.今後は,テヅルモヅルのもっとディープな部分を掘り下げていきたいと思っています.
今後も,益々研究に励んで参ります.

イボサキワレテヅルモヅル(2014/1撮影
@串本海中公園)