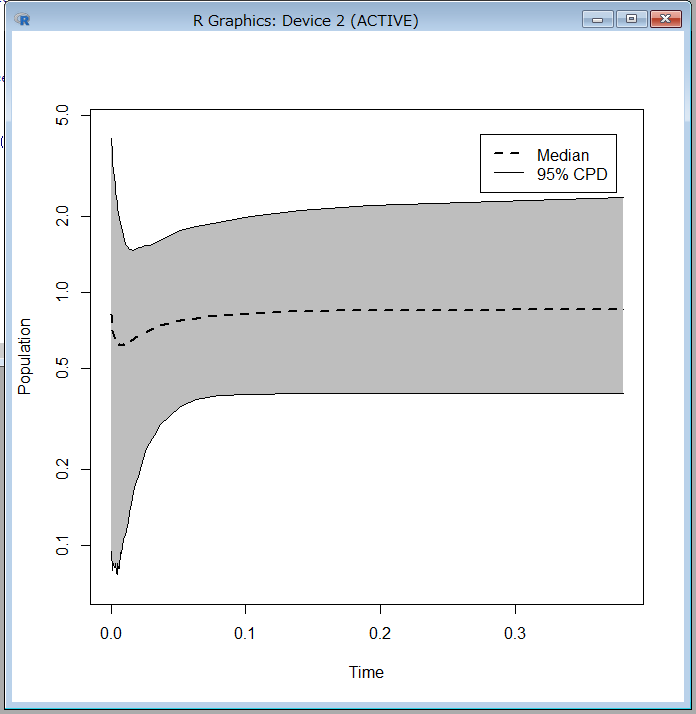
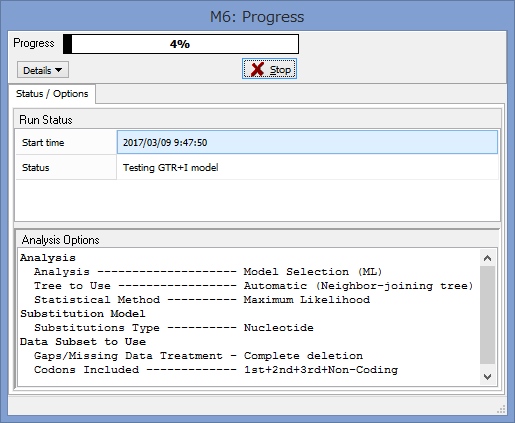
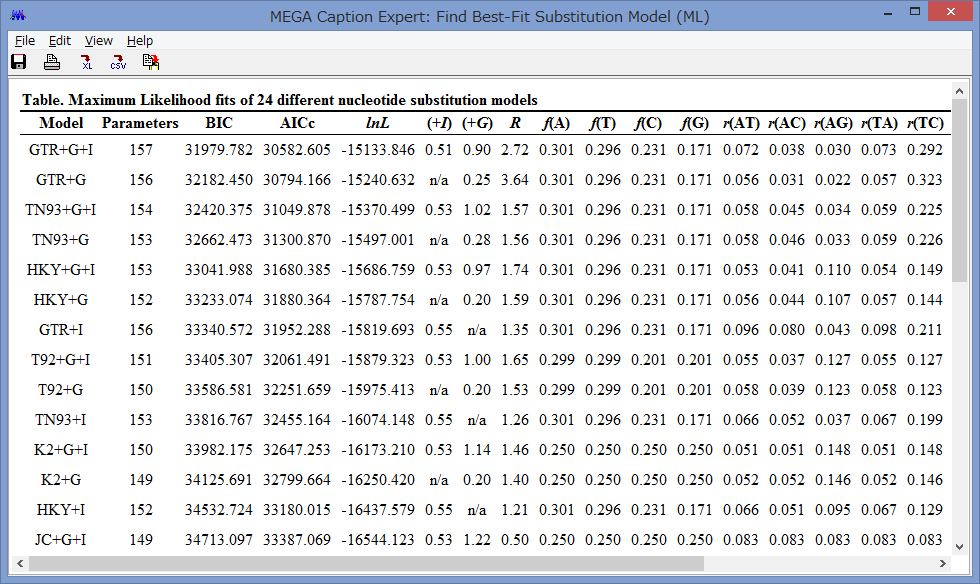
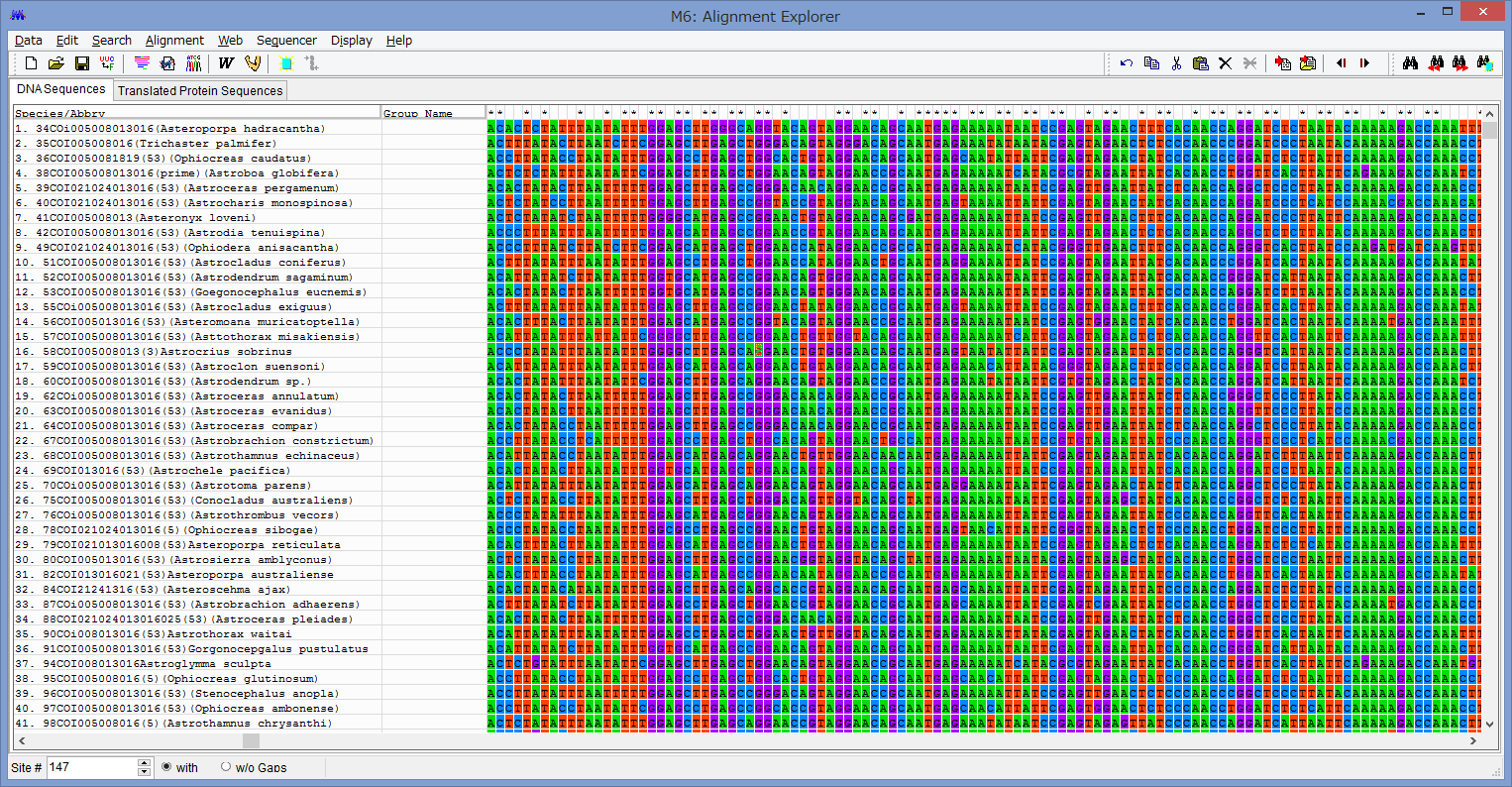
2/1に講演をします。以下詳細です。
—————————————————–
よさこい生態学セミナー
【日 時】2019年2月1日(金)16時30分~18時00分
【参加費】無料
【会 場】高知大学物部キャンパス 附属暖地フィールドサイエンス教育研究センター講堂
http://www.kochi-u.ac.jp/outline/campus_map_monobe.html
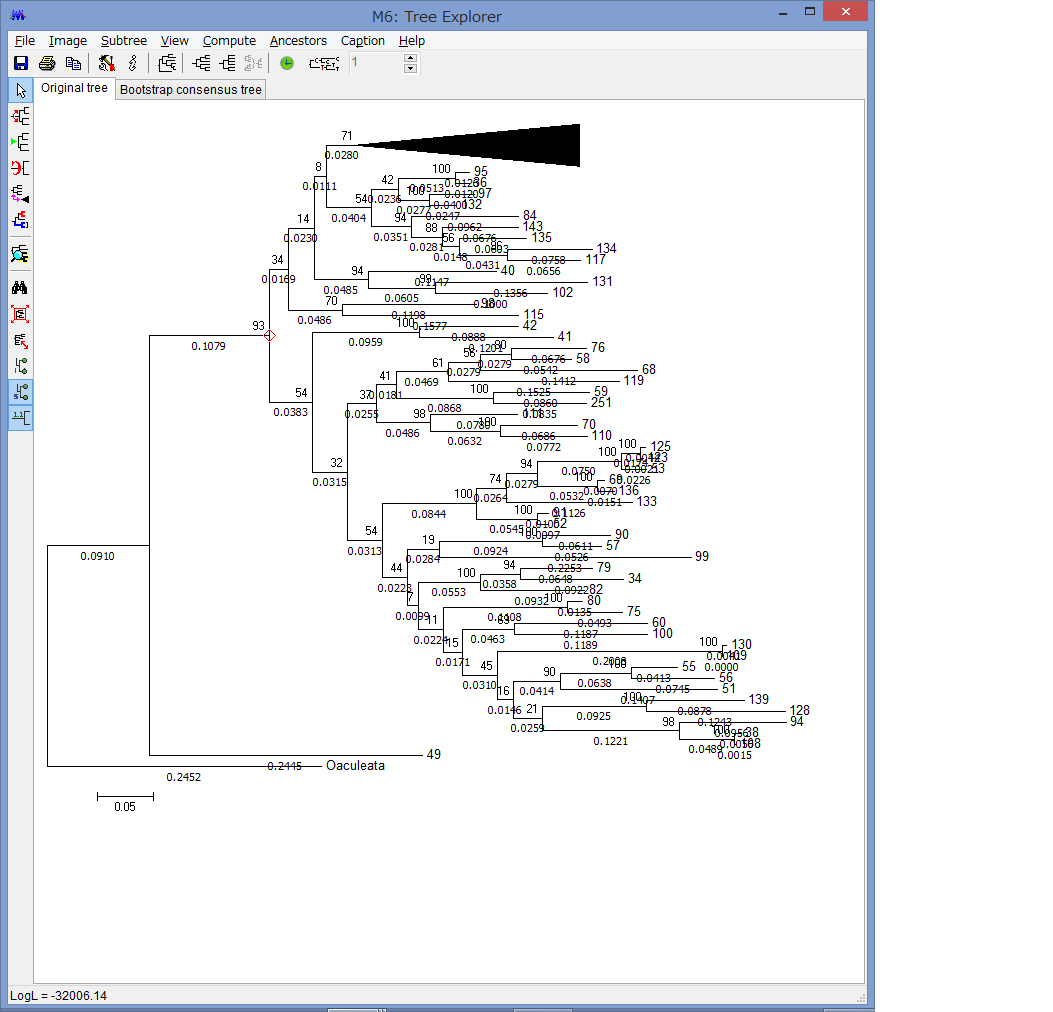
「クモヒトデを研究する
~フィールドワークに基づくアプローチ」
—————————————————–
参加無料のようです。要旨など、詳しくは以下のHPをご参照ください。
http://yosakoiseminar.blogspot.com/
実家にとても近い場所で凱旋?講演です。
お声をかけてくださった高知大学の鈴木紀之先生、
ありがとうございます。
頑張ります!