MEGAは視覚的にわかりやすいGUI形式ですので,
特に解説は必要ないと思いますが,備忘録的に.
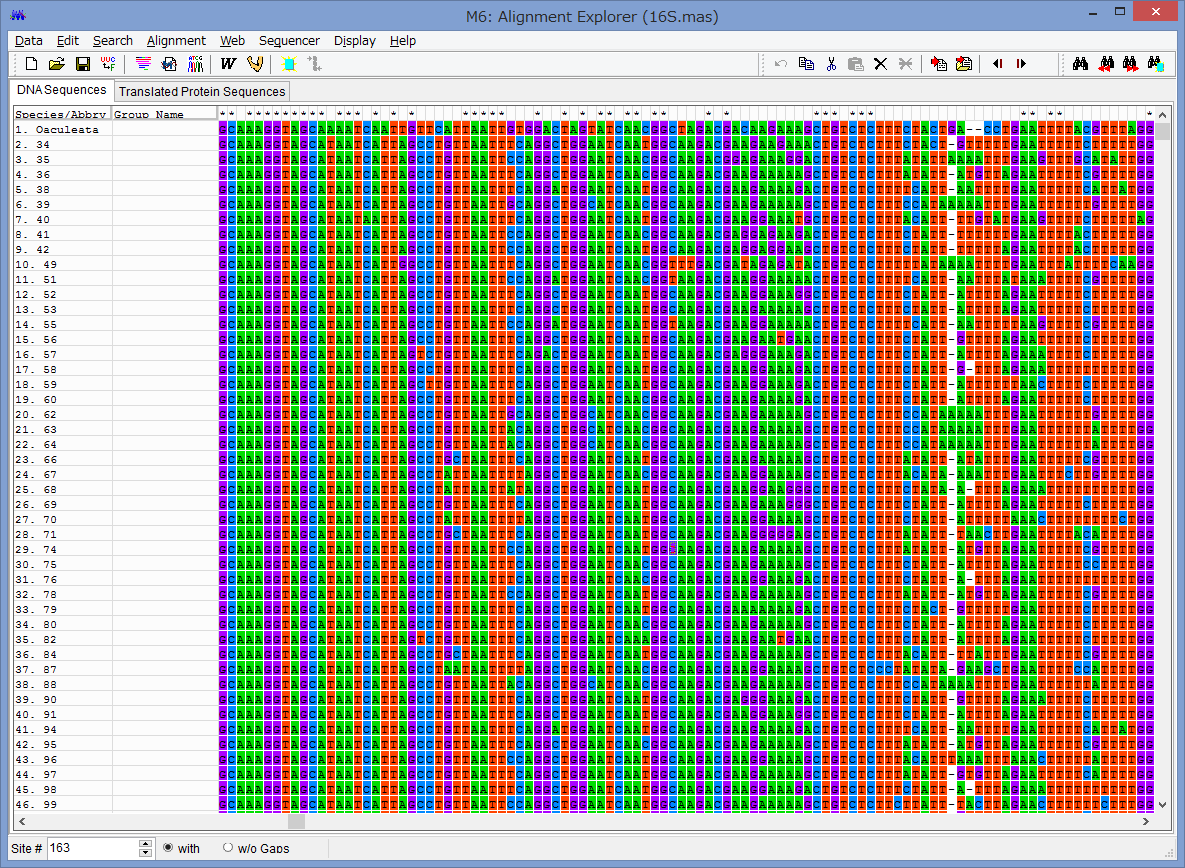
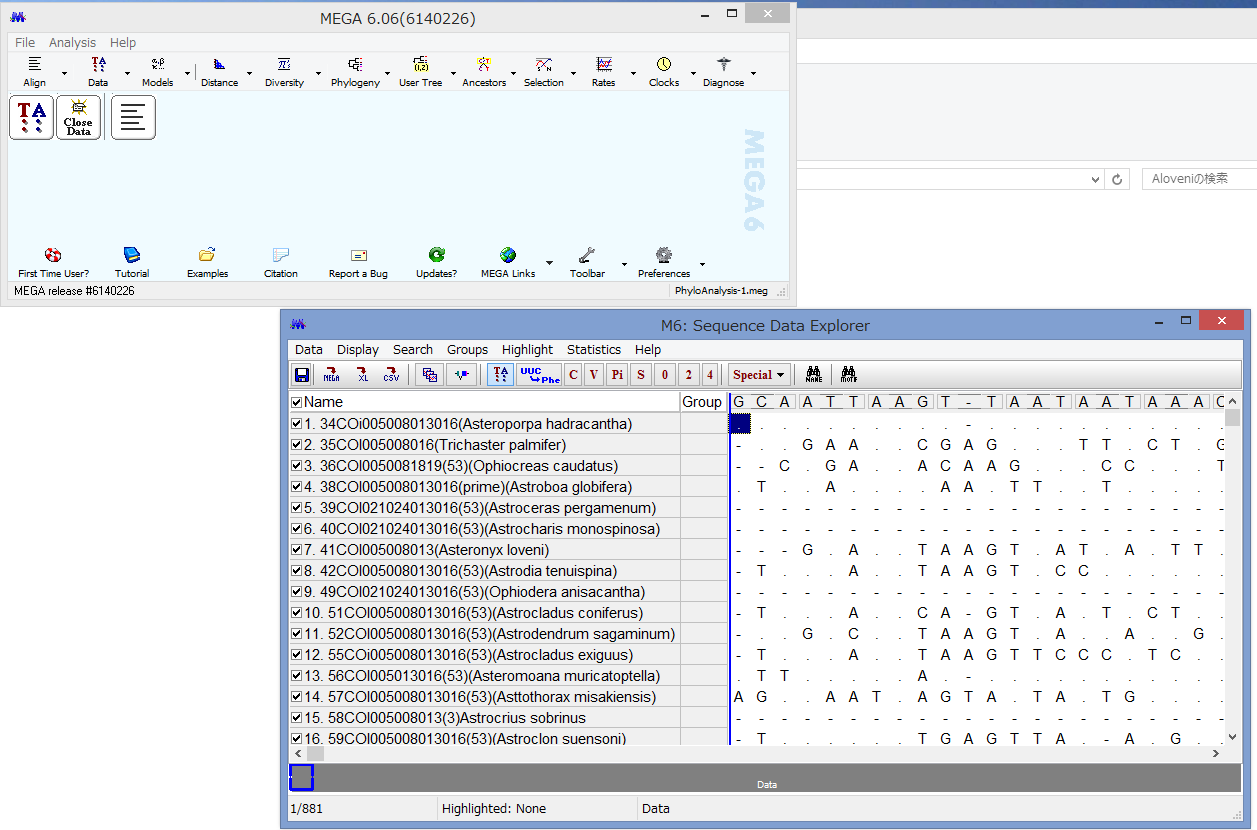
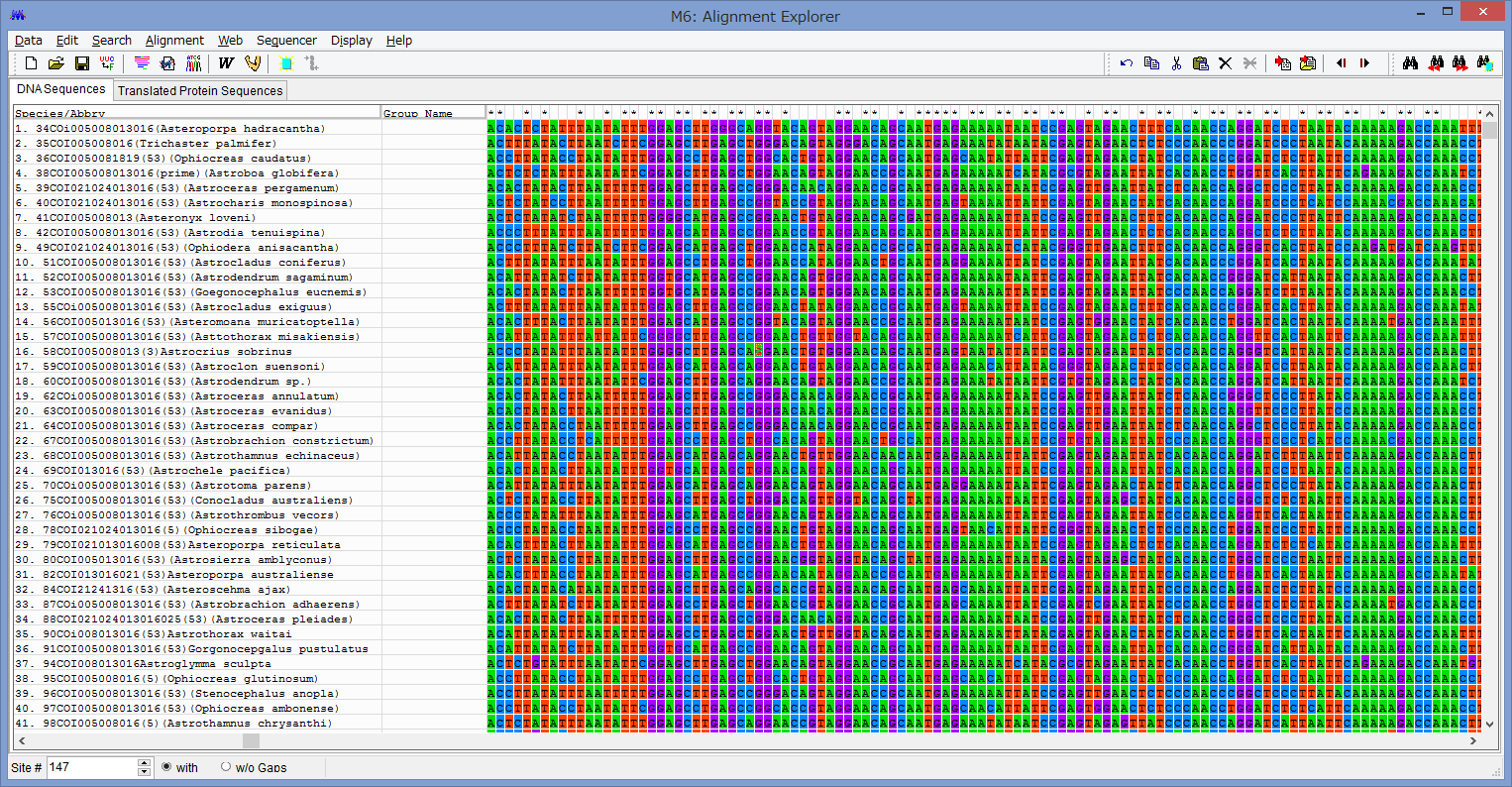
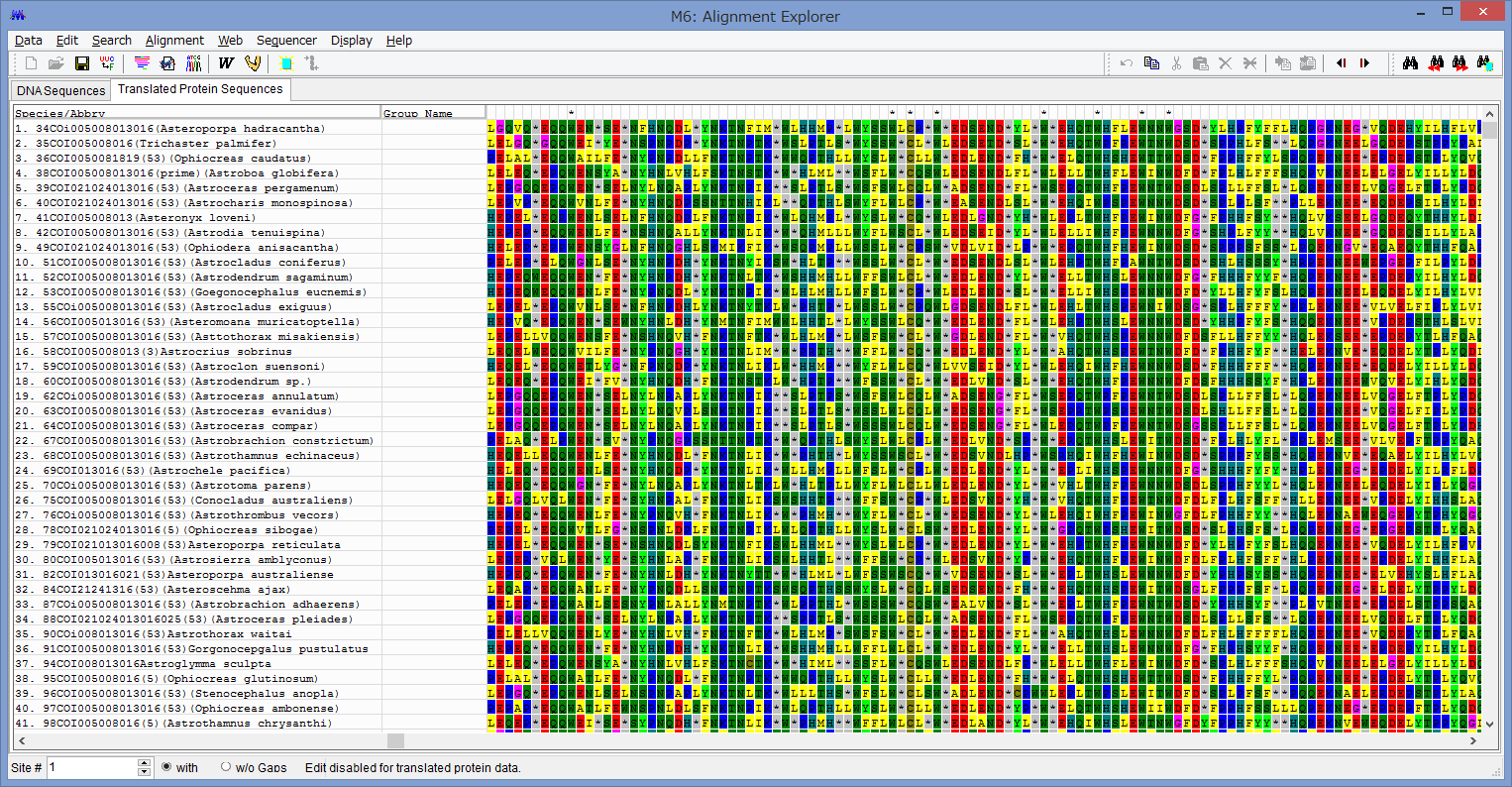
アライメントした配列を用意します.COI配列です.
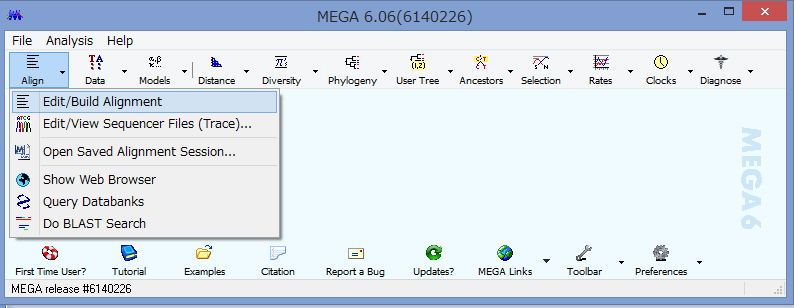
Data→Phylogenetic Analysisを選択.
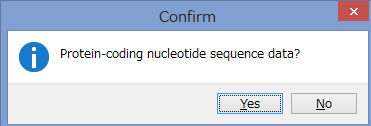
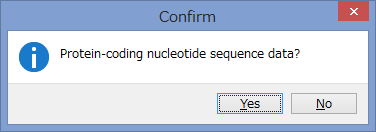
アミノ酸のコーディング領域かどうかを聞かれます.
今回はCOIなのでYesです.

うまくいけばこのようなウィンドウが表示されます.
MEGAの基本ウィンドウ(ここではMEGA 6.06(6140226))でPhylogeny→Construct/Test Maximum Likelihood Tree…を選択.
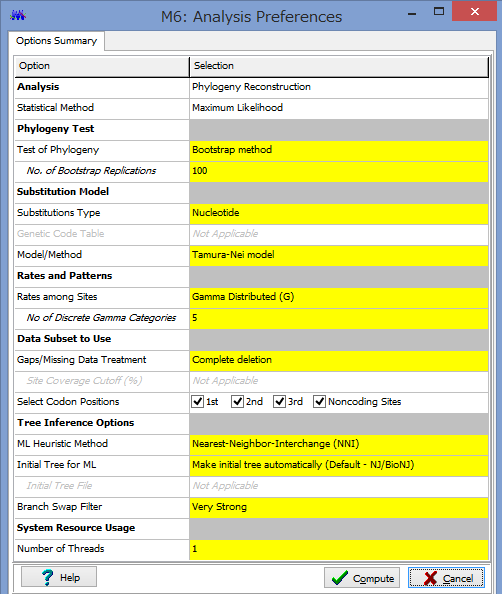
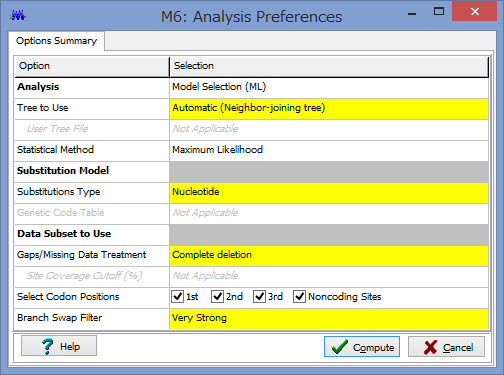
ここで解析のパラメーターを設定します.
Phylogeny Test :どのように系統を検定するかです.各枝の信頼性が得たい場合はここでBootstrap Methodを選択しましょう.
No. of Bootstrap Replications:Bootstrap検定を何回行うかです.100回でも構いませんが,1000回くらいはしたほうが無難です.2-3000回やってる論文もたまに見かけます.
Substitution Type:塩基(Nucleotide)かアミノ酸(Amino Acid)が選べます.選べないと困ります.
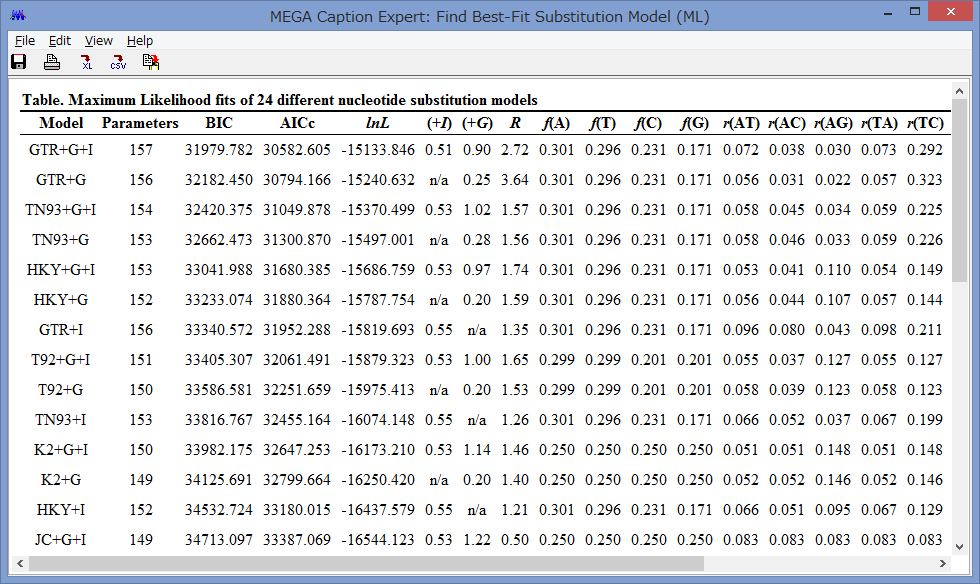
Model/Method:塩基(アミノ酸)置換モデルが選べます.
Rates among Sites:座位ごとの置換の頻度の違いを,ガンマ分布に基づいて分類するか否かを設定できます.
No of Discrete Gamma Categories:ガンマカテゴリ数を設定できます.
Gaps/Missing Data Treatment:ギャップの扱いの設定です.Complete deletionとすると,一つでもギャップがあるサイト(列)を解析からのぞけます.
ML Heuristic Method:最尤法系統樹の探索方法を選べます.系統樹探索方法についてはまたどこかで書くかもしれませんが,局所解がたくさんあるようなデータセットだと,ここでの方法選びは結構重要だと思います.いくつか試してみても良いかもしれません.
Initial Tree for ML:系統樹探索を行う際の,最初の出発点の系統樹の作成法を選びます.上記した通り,これも結構重要だと思います.デフォルトのNJでも問題はないと思いますが,もし既にそれらしい系統樹があるのであれば,それを設定する事もできます.
Blanch Swap Filter:系統樹探索の際の枝の入れ替えの大胆さを決めます.Strongにすると枝長の入れ替えがより消極的になり,解析時間は短くなりますが,考慮する系統樹は少なくなります.よりWeakにすると,枝長の入れ替えが大胆になり,解析時間は長くなりますが,考慮する系統樹が多くなるようです.
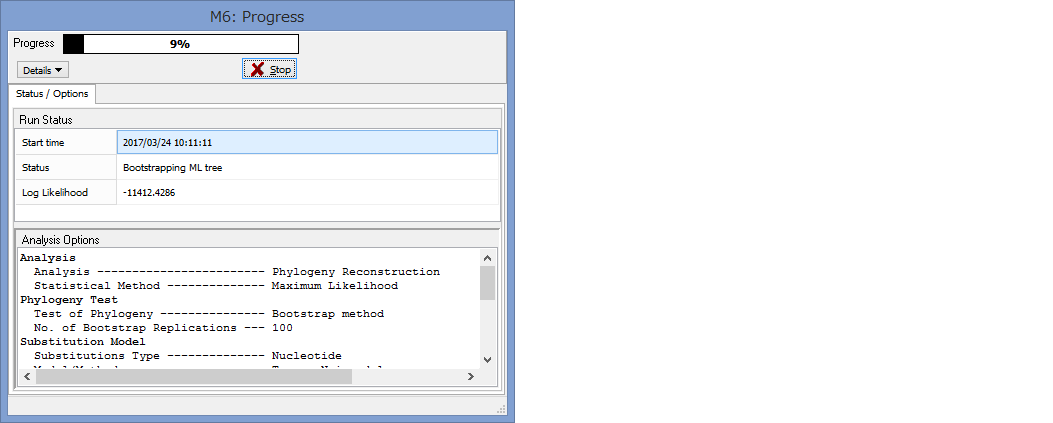
パラメーター設定が終わったら,Computeをクリックして解析開始です.
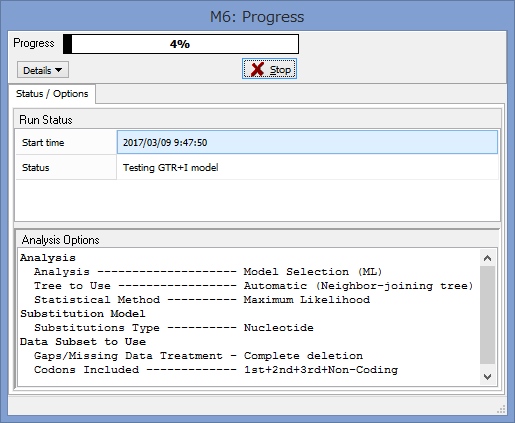
Progressが100%になるまで,気長に待ちましょう.
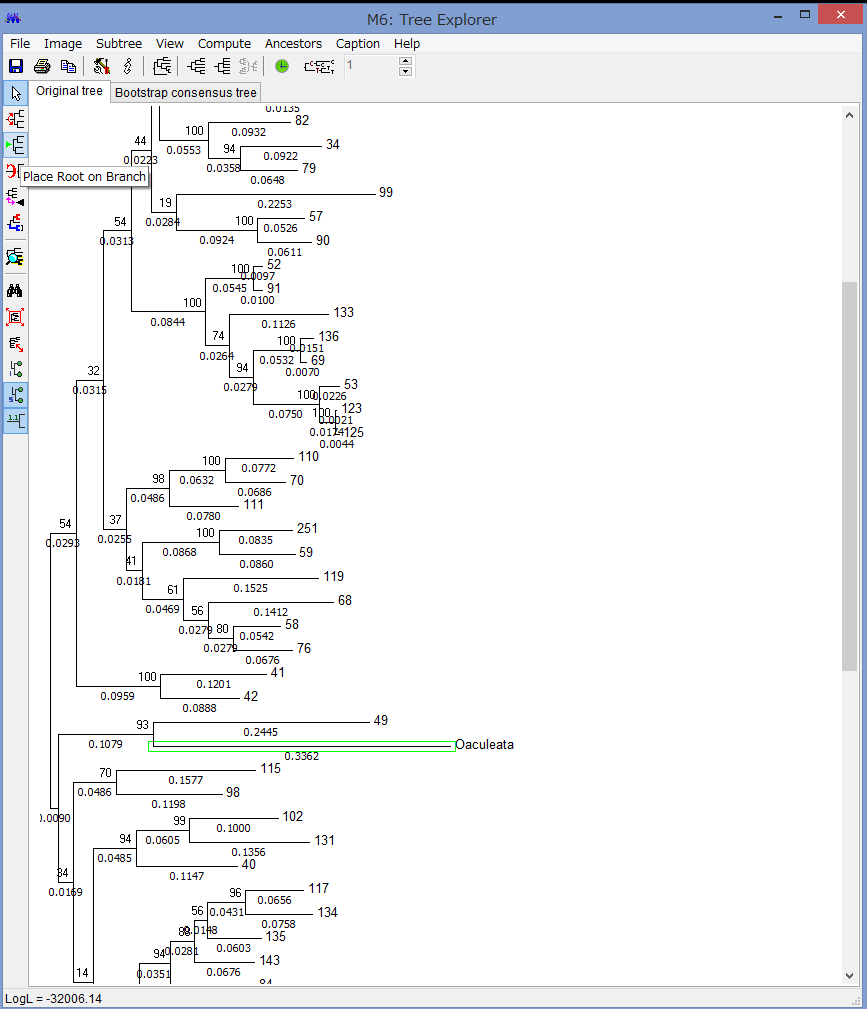
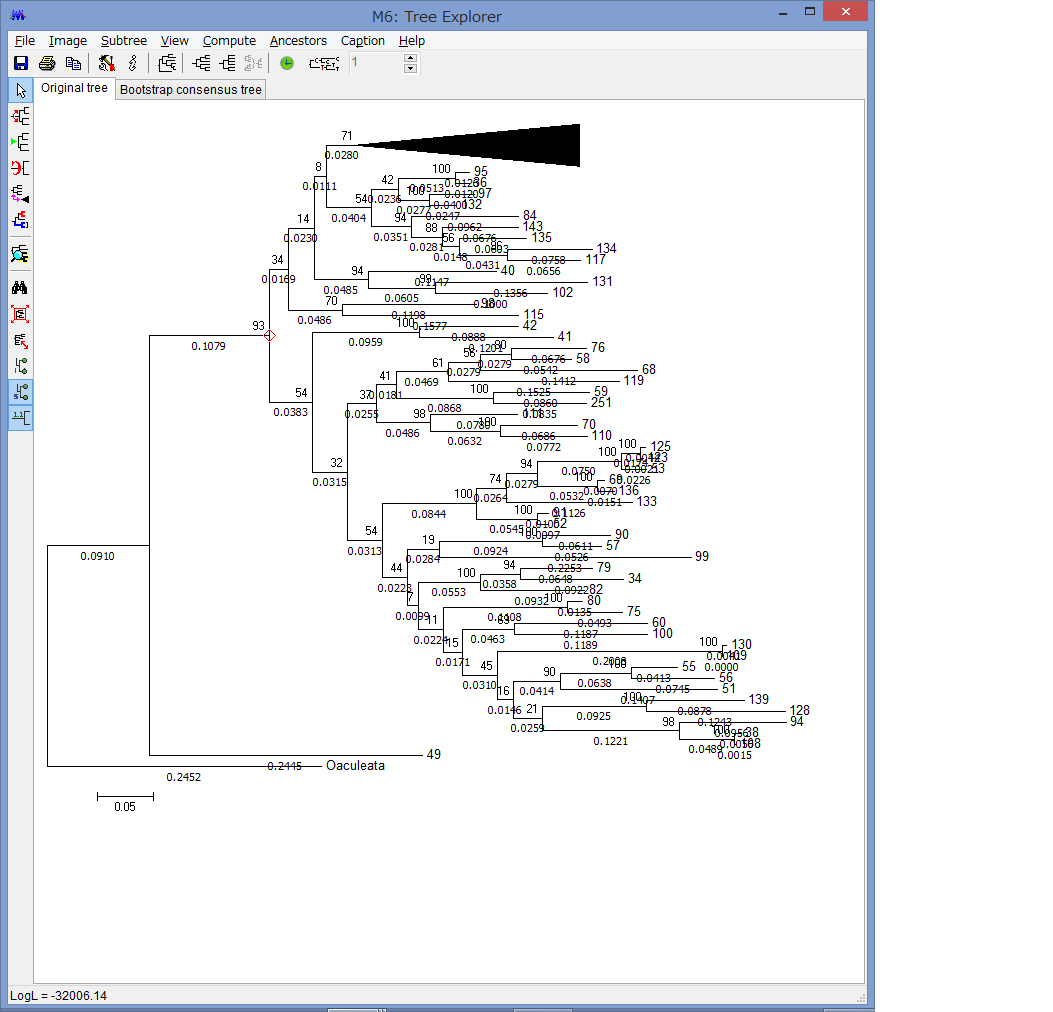
解析が終わると,このようにTree Explorerに系統樹が表示されます.
各枝の上にブートストラップ確率(2桁の整数)と枝の下に枝長(有理数)が示されています.
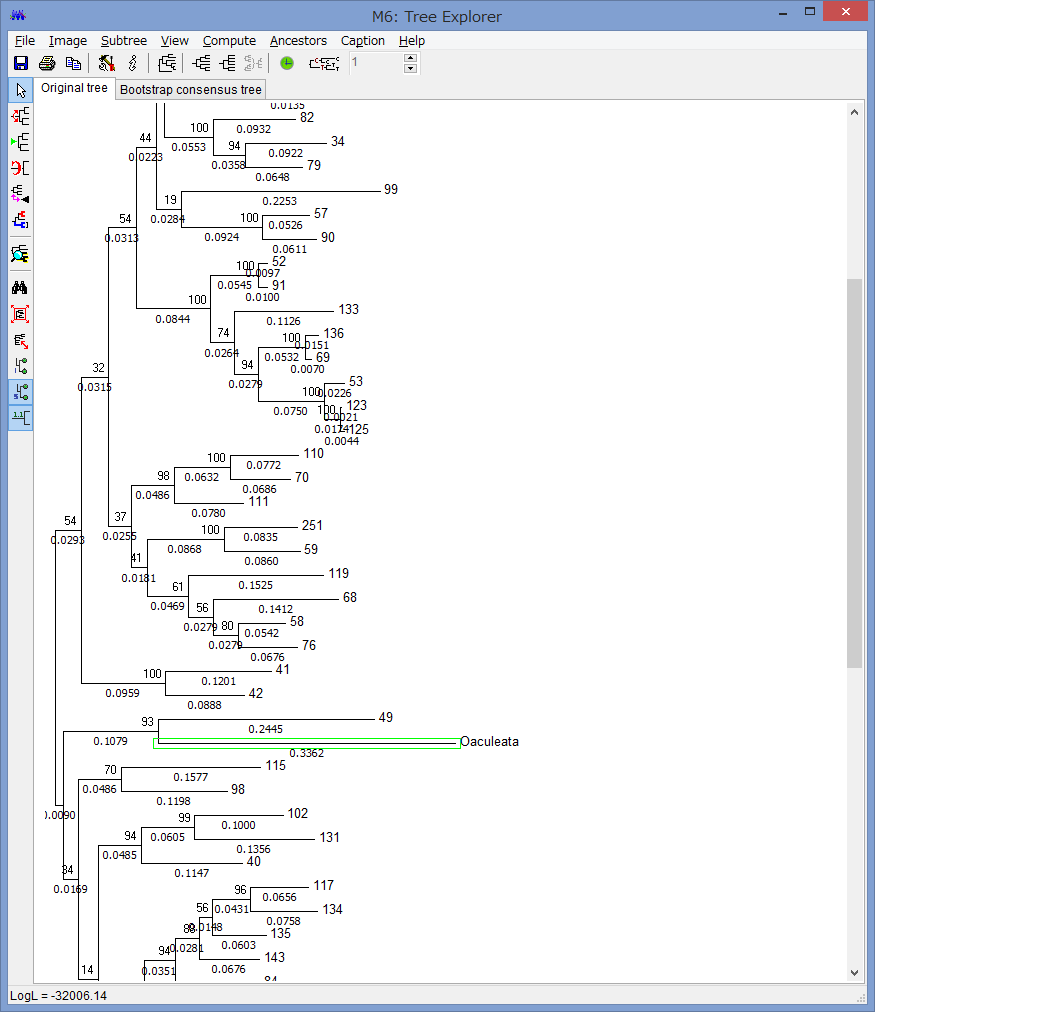
このExplorerで色々系統樹をいじれます.例えば特定の枝を選択して,左上のコマンドの中からPlace root on Branchを選ぶと
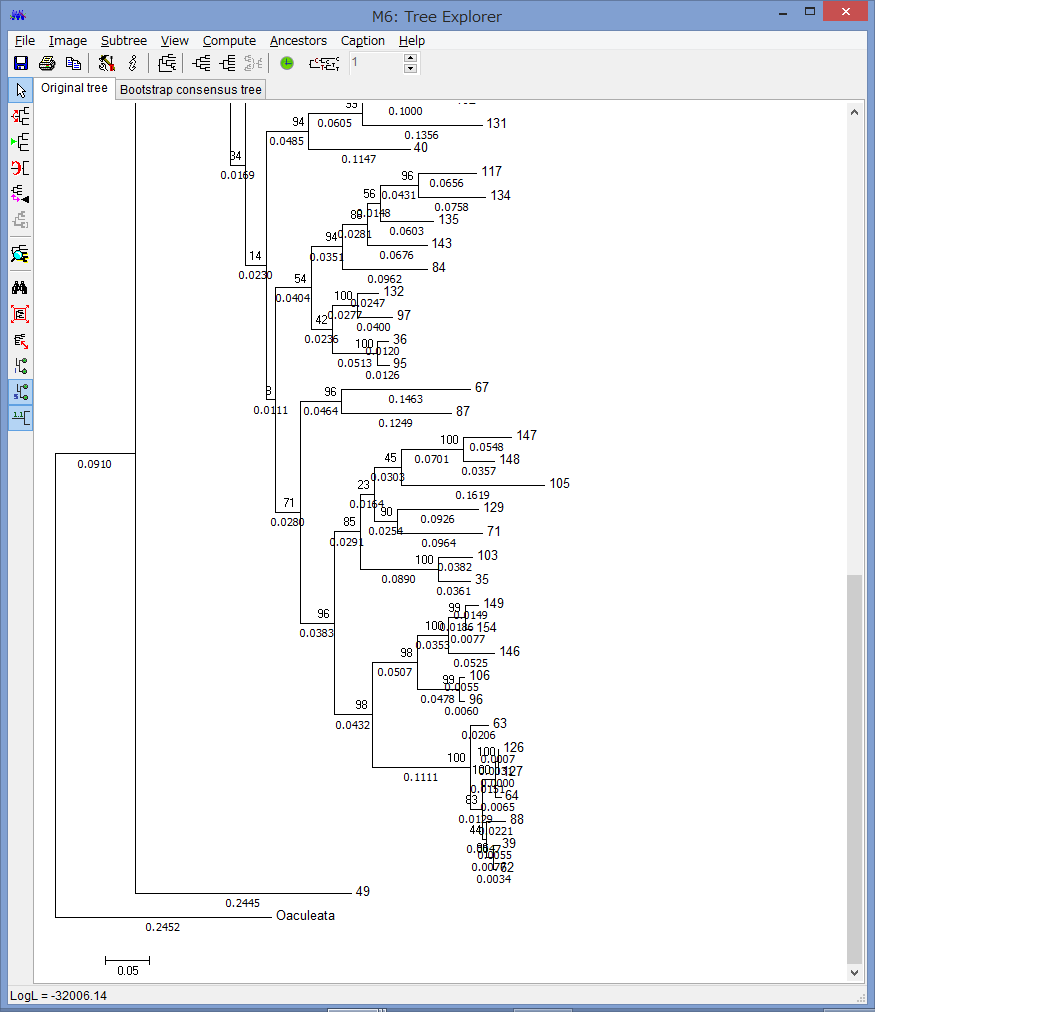
外群を指定できます.
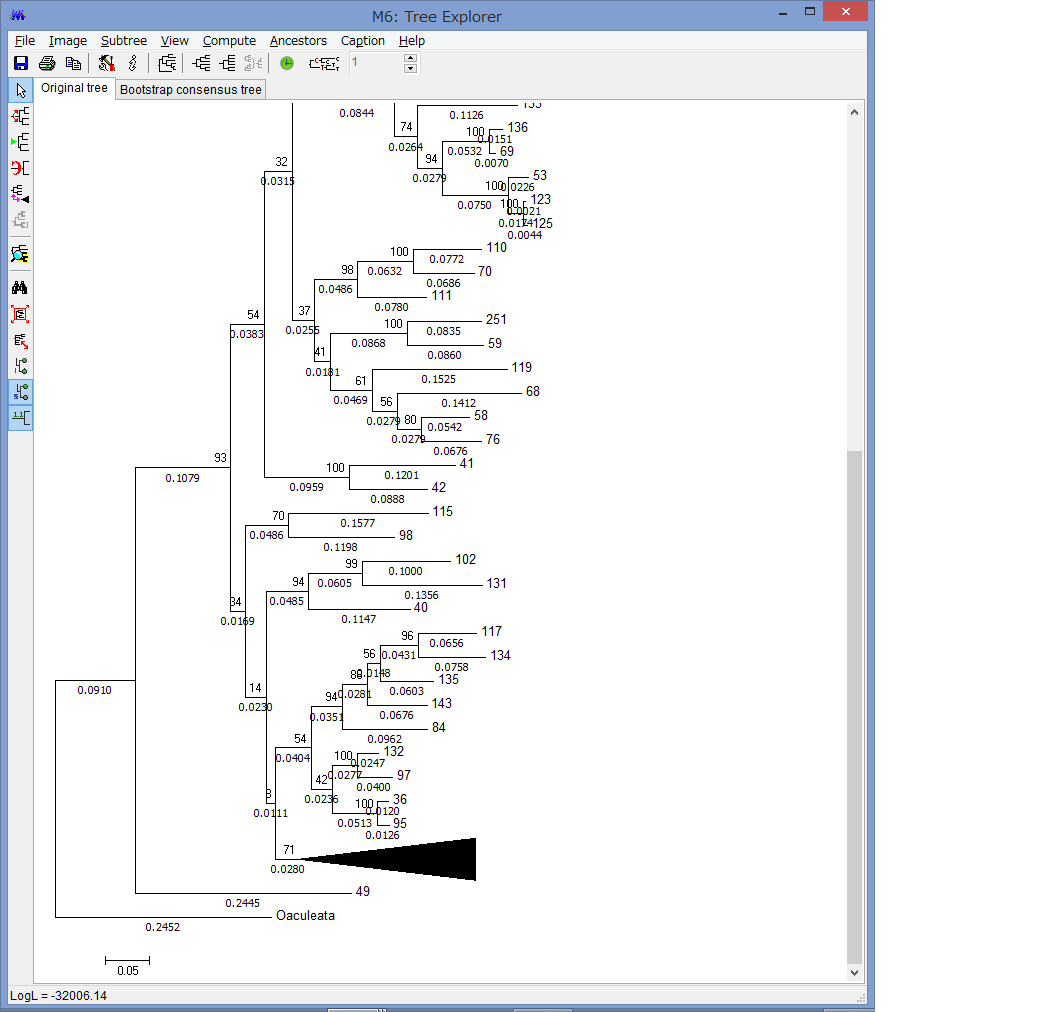
他にもCompress/Expand Subtreeで,枝を一つにまとめたり,

Fit Tree to Screenで,ウィンドウ内に系統を収めたり,
Flip Subtreeで枝の上下を入れ替えたりできます.
良い時代になりました.
この他にもたくさんのコマンドがありますが,感覚的に理解できると思いますので,いろいろ試してみてください.
今日はここまで.